
Cancer Genomic instability
-
Genomic instability as a major driving force of tumorigenesis
-
Cancer development is a complex multi-step process, involving a wide network of cellular events. Genomic instability, a hallmark of cancer, enables the acquirement of new characteristics required for tumorigenesis. We focus on the different cellular pathways which are altered under aberrant oncogene expression, causing various cellular stress reactions leading to perturbed DNA replication dynamics and chromosomal instability. We have shown that aberrant oncogene over-expression forces DNA replication to proceed under suboptimal conditions such as nucleotide deficiency, leading to replication stress and DNA damage (Bester et al. Cell 2011). Recently, we have demonstrated that activation of the RAS oncogene perturbs DNA replication due to increased topoisomerase 1 expression, disturbing the regulation of R-loop homeostasis and resulting in genomic instability (Sarni et al. Cell Reports 2022).
Another aspect of our work focuses on the tumor suppressor gene TP53, known as the “guardian of the genome” as it plays a key role in restricting tumor initiation and progression. We study the mechanisms underlying genomic instability including the catastrophic chromothripsis instability event, associated with aggressive tumorigenesis in cells deficient for wild type p53. We found that p53 loss leads to replication stress resulting in genomic instability. We found that nucleotide supplementation rescues replication stress, DNA damage and proliferation crisis (in progress).
Altogether, unfolding the molecular basis underling genome instability in cancer may open new potential prevention and therapeutic approaches.
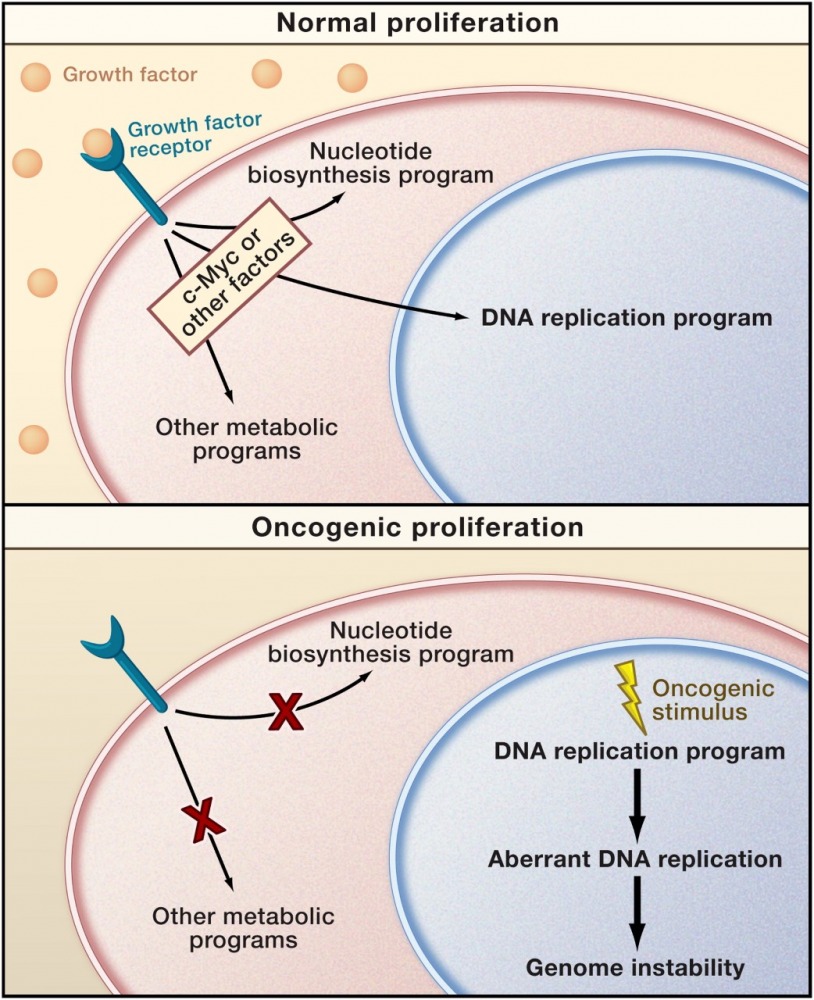
A mismatch between proliferation and metabolite production may characterize oncogenic cell cycles (Venkitaraman 2011, Preview in Cell on Bester et al., Cell 2011).
-
Recurrent chromosomal breakage and cancer
-
Common fragile sites (CFSs) are genomic regions susceptible to replication stress and are hotspots for chromosomal instability in cancer. CFSs instability accelerates tumorigenesis by increasing the probability of acquiring genomic alterations, supporting cancer progression. We aim to understand the features underlying their sensitivity during transformation.
We have previously shown that the repertoire of fragile sites is dynamic and dependent on the replication stress inducer. We demonstrated that the replication inhibitor Aphidicolin and various oncogenes lead to a different landscape of recurrent fragility, reflecting the variability in genomic alteration among tumors overexpressing different oncogenes (Miron et al. Nature Communications 2015).
Recently, we explored the transcriptional profile, DNA replication timing and the 3D genome organization under aphidicolin treatment and overexpression of various oncogenes. Analyzing the aphidicolin-induced fragility revealed a fragility signature, comprised of a topologically associated domain (TAD) boundary overlapping a highly transcribed large gene with replication timing delay. CFS stability may be compromised by incomplete DNA replication and repair in TAD boundaries core fragility regions leading to genomic instability (Sarni et al. Nature Communication 2020).
Currently, we are studying the mechanisms underlying oncogene induced fragility. Our preliminary results indicate the role of transcription perturbation in fragility (on-going projects). Our goal is to establish a strategy that can predict which pathways are disrupted in a given cancer type according to its chromosomal breakpoint pattern.
Altogether, identifying the various fragility signatures will allow a more comprehensive understanding of the role of CFSs in cancer development and will highlight the various mechanisms promoting genomic instability in cancer.

Delayed replication of highly transcribed large genes in TAD boundaries leads to DNA breaks.



